INTRODUCTION
Les antiviraux agissent sur des cibles précises qui peuvent être virales (enzymes virales) ou plus rarement cellulaires (récepteurs du virus). Dans les deux cas, c’est le plus souvent la variabilité génétique des virus qui va conduire à la résistance. Les virus mutants présentent une diminution de sensibilité (= résistance) et sont sélectionnés en présence de l’antiviral concerné. Les traitements au long cours favorisent l’émergence de virus résistants.
Cliniquement, la résistance virale est caractérisée par l’absence d’efficacité clinique d’un traitement antiviral. Virologiquement, elle est mise en évidence par une réplication virale (charge virale) persistante sous traitement.
ORIGINE DE LA RÉSISTANCE AUX ANTIVIRAUX
Trois éléments interviennent dans la résistance aux antiviraux : le virus, l’antiviral, le patient.
Le virus peut être naturellement résistant (virus sauvage, non muté) mais généralement il devient résistant par l’acquisition de mutations de résistance (virus muté). Ces mutations aléatoires sont dues aux erreurs que génèrent les ADN (acide désoxyribonucléique) ou ARN (acide ribonucléique) polymérases virales lors de la réplication.
En fonction de l’antiviral, la résistance apparaît après un nombre plus ou moins élevé de mutations. Un virus a une barrière génétique faible pour un antiviral lorsqu’une mutation du virus suffit à conférer la résistance. À l’opposé, lorsqu’un virus doit accumuler plusieurs mutations pour être résistant, la barrière génétique du virus pour l’antiviral est dite élevée.
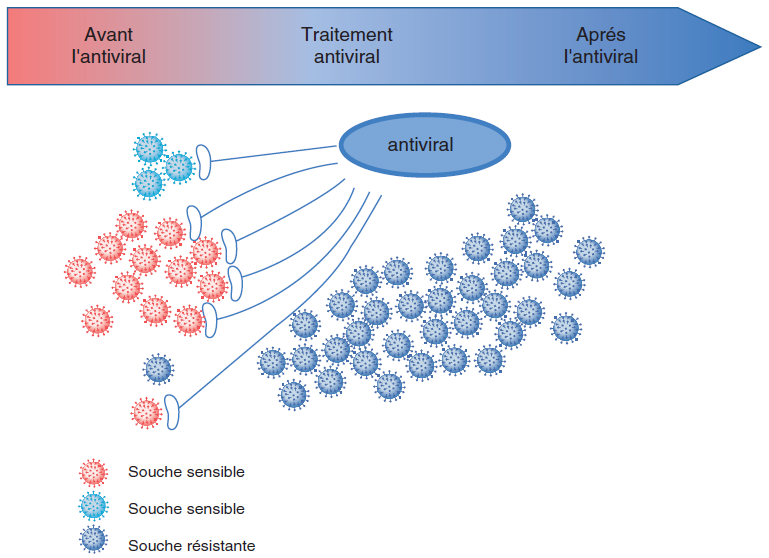
Un virus sensible a une capacité réplicative (Fitness) maximale en l’absence d’antiviral. En présence d’antiviral, le virus résistant a une capacité réplicative supérieure au virus sensible. En l’absence d’antiviral, c’est l’inverse, le virus sensible a généralement une capacité réplicative supérieure à celle du virus résistant (figure IV.4.1).
Pour de nombreux virus, il faut également prendre en compte la diversité naturelle de la population virale présente chez l’individu infecté (ou quasi-espèce). L’efficacité de l’antiviral peut dépendre de la prévalence et de l’incidence des mutations associées à la résistance dans la quasi-espèce.
L’antiviral a naturellement une puissance variable sur l’inhibition de la réplication virale. Les traitements antiviraux doivent être les plus puissants possibles et sont donc souvent utilisés en combinaisons (multithérapies avec cibles différentes). La concentration de l’antiviral et sa métabolisation peuvent également varier, par exemple en fonction de la posologie administrée ou des interactions médicamenteuses (ou alimentaires) qui peuvent rendre les concentrations d’antiviraux insuffisantes et inefficaces.
Le patient a une influence directe sur le niveau de réplication du virus (facteurs génétiques, immunologiques et comportementaux) et sur les concentrations de l’antiviral (observance du traitement, métabolisme). Les effets indésirables des antiviraux sont souvent à l’origine d’un défaut d’adhérence du patient à son traitement.
La résistance est le plus souvent la conséquence d’un défaut d’adhérence au traitement.
MÉCANISMES DE RÉSISTANCE
MODIFICATION DE LA CIBLE
La majorité des antiviraux ont pour cible des enzymes virales (polymérases, protéases, intégrases, phosphotransférases, terminases…). L’antiviral mime par sa structure le substrat de l’enzyme virale et conduit à un arrêt ou un dysfonctionnement de cette dernière. La résistance à l’antiviral est la conséquence d’une mutation sur le gène codant l’enzyme et aboutissant à sa modification. Cette modification entraîne un défaut de reconnaissance des antiviraux par le site catalytique de l’enzyme :
▶ polymérases : analogues nucléosidiques et nucléotidiques, analogues du pyrophosphate, inhibiteurs spécifiques des polymérases ;
▶ protéases : antiprotéases ;
▶ intégrase du VIH-1 : anti-intégrases ;
▶ phosphotransférase du CMV (cytomégalovirus) : antiphosphotransférase (maribavir) ;
▶ terminases du CMV : antiterminases (letermovir) ;
▶ neuraminidase des virus Influenza : inhibiteurs de la neuraminidase.
Parfois la cible de l’antiviral n’est pas une enzyme mais un élément de la structure du virus. Dans ce cas, la mutation modifie cette structure qui n’est plus reconnue par l’antiviral ce qui conduit à :
▶ un défaut de fixation sur l’élément structural, ex. mutation de la Gp41 du VIH-1 conduisant à un défaut de fixation de l’enfuvirtide et à un défaut d’inhibition de la fusion ;
▶ un blocage de l’élément structural, ex. mutation de la protéine M2 du virus Influenza A conduisant à un blocage des canaux ioniques, à un défaut d’acidification et un défaut de la décapsidation.
Certains antiviraux ont pour cible un récepteur cellulaire. Dans ce cas, la mutation restaure la capacité de fixation de l’antiviral à la cible, en présence de l’antiviral :
▶ adaptation du virus à la cible en présence de l’antiviral, ex. :
– le VIH-1 modifie sa Gp120 pour reconnaître son corécepteur (le CCR5) malgré la fixation du maraviroc,
– le VIH-1 augmente l’affinité de la Gp120 pour le CCR5 ce qui permet de déplacer le maraviroc ;
▶ capacité du virus à trouver une nouvelle cible, ex. sélection de populations de VIH utilisant le corécepteur CXCR4.
MODIFICATION DE LA MÉTABOLISATION DES PRODROGUES D’ANTIVIRAUX
Les analogues nucléosidiques et nucléotidiques doivent être triphosphorylés pour être reconnus par le site catalytique des polymérases. Une des 3 phosphorylations est réalisée par les kinases virales (thymidine kinase ou phosphotransférases virales pour l’aciclovir et le ganciclovir).
Une mutation dans le gène de la kinase virale peut aboutir à une diminution de l’activité ou de la spécificité de l’enzyme conduisant à une absence de phosphorylation de l’antiviral et à son inactivité.
Les analogues nucléotidiques possèdent d’emblée la phosphorylation réalisée par la kinase virale et ne nécessitent donc que les phosphorylations réalisées par les kinases cellulaires. Dans ce cas, c’est la modification de l’activité de phosphorylation cellulaire (génétique, compétition) qui peut aboutir à l’inefficacité de l’antiviral.
MÉCANISMES MULTIFACTORIELS
L’interféron alpha est un antiviral naturellement produit par l’organisme humain en cas d’infection virale. Il induit l’expression de plus de 200 gènes humains conduisant à une inhibition de la réplication virale par de nombreux mécanismes. Les cellules exposées à l’interféron sont réfractaires à l’infection essentiellement par réduction de la durée de vie des ARNm viraux et de leur traduction.
Il existe des résistances naturelles et acquises à l’interféron alpha, mais les déterminants sont très mal connus et probablement multifactoriels.
C’est également le cas de la résistance à la ribavirine. En effet, la ribavirine a des mécanismes d’action multiples : inhibition des ARN polymérases ARN dépendantes virales, déplétion du pool de dNTP (désoxyribonucléoside triphosphate), effet mutagène létal, effet immunologique, etc. ce qui rend difficile la caractérisation de la résistance des virus à cet antiviral.